

Genomics workflows, Part 7: analyze public RNA sequencing data using AWS HealthOmics
Genomics workflows process petabyte-scale datasets on large pools of compute resources… In our use case, life science research teams use workflows written in Nextflow to process RNA-Seq datasets in FASTQ file format… For example, scientists might need to do the following: Manually download …
Genomics workflows process petabyte-scale datasets on large pools of compute resources. In this blog post, we discuss how life science organizations can use Amazon Web Services (AWS) to run transcriptomic sequencing data analysis using public datasets. This allows users to quickly test research hypotheses against larger datasets in support of clinical diagnostics. We use AWS HealthOmics and AWS Step Functions to orchestrate the entire lifecycle of preparing and analyzing sequence data and remove the associated heavy lifting.
Use case
In genomics, transcription relates to the process of making a ribonucleic acid (RNA) copy from a gene’s deoxyribonucleic acid (DNA). Usually, RNA is single-stranded, although some RNA viruses are double-stranded. With RNA sequencing (RNA-Seq), scientists isolate the RNA, prepare an RNA library, and use next-generation sequencing technology to decode it. Organizations around the world use RNA-Seq to support clinical diagnostics.
In our use case, life science research teams use workflows written in Nextflow to process RNA-Seq datasets in FASTQ file format. Following their initial RNA-Seq studies on internal datasets, scientists can extend their insights by using public datasets. For example, the Gene Expression Omnibus (GEO) functional genomics data repository is hosted by the National Center for Biotechnology Information (NCBI) and offers multiple download options and formats. Scientists can download datasets in FASTQ format from GEO File Transfer Protocol (FTP) and compress them into the .gz format before further analysis.
Scaling and automating the data ingestion can be challenging. For example, scientists might need to do the following:
- Manually download FASTQ files and invoke their analysis pipelines
- Monitor the workflow runs, which can span hours, days, or weeks
- Manage the infrastructure for performance and scale
This blog post presents a solution that removes this undifferentiated heavy lifting.
Prerequisites
To build this solution, you must be analyzing transcriptomic sequencing data with the Nextflow workflow system and make use of GEO FASTQ datasets. In addition, you must do the following:
- Create three Amazon Simple Storage Service (Amazon S3) buckets with the following purposes:
- Uploaded GEO Accession IDs (GEO IDs)
- Ingested FASTQ datasets
- RNA-Seq output files
- Create one Amazon DynamoDB table to track the status of data ingestion. This helps with checkpointing and avoids repetitive ingestion jobs so that you can keep data ingestion cost to a minimum.
Solution overview
Using AWS, you can automate the entire RNA-Seq Nextflow pipeline. Users only need to provide the GEO IDs, then the pipeline ingests the corresponding FASTQ sample files and performs the subsequent data analysis.
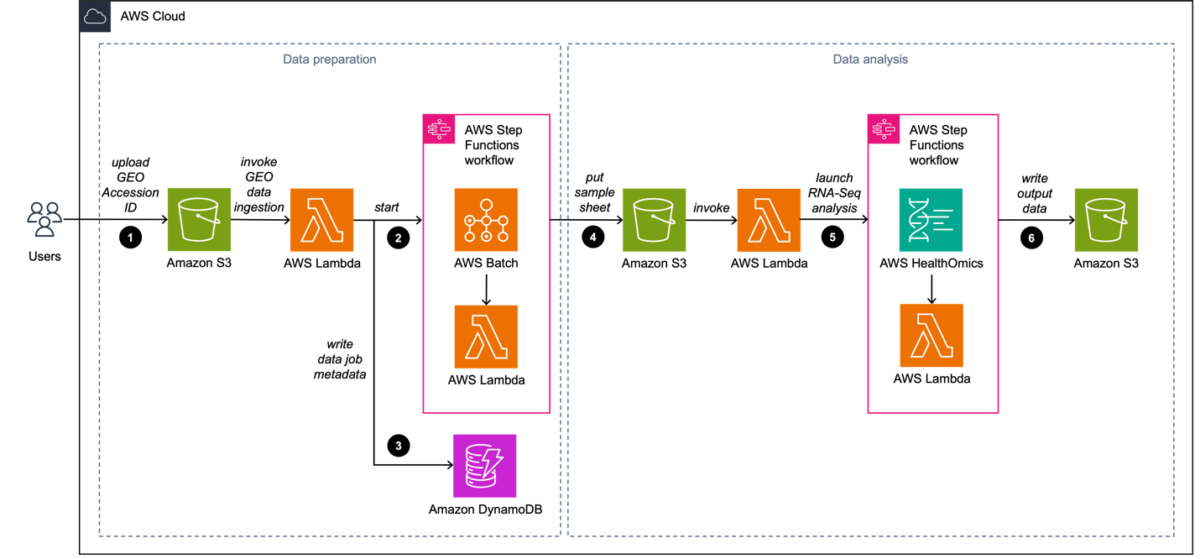
Our solution, shown in Figure 1, uses a combination of AWS HealthOmics and AWS Step Functions. HealthOmics manages the compute, scalability, scheduling, and orchestration required for processing large RNA-Seq datasets. This helps scientists focus on writing their pipelines in Nextflow while AWS takes care of the underlying infrastructure. Step Functions adds reliability to the workflow from dataset ingestion to output archival. Automating the entire workflow also helps with tracing specific invocations and troubleshooting errors.

Figure 1. RNA sequencing using HealthOmics
Our solution includes the following:
- The scientist creates and uploads a CSV file to the GEO metadata S3 bucket. The CSV file includes a reference to the specific GEO ID that is ingested. An Amazon S3 Event Notification configured on s3:ObjectCreated events (in this case, the CSV file upload) invokes an AWS Lambda function.
- The Lambda function first extracts the corresponding Sequence Read Run (SRR) IDs of the GEO ID. Next, it starts a Step Functions state machine with the following input parameters: the SRR IDs, species of the samples, and GEO ID. The state machine uses an AWS Batch job queue for parallel ingestion.
- The Lambda function writes the following metadata to a DynamoDB table for future reference:
- Ingested GEO ID and corresponding list of SRR IDs
- Amazon S3 output paths to the ingested FASTQ files
- Overall workflow status
- Ingested species
- Upon ingestion completion, the state machine puts the RNA-Seq sample sheet into the FASTQ S3 bucket. This invokes a Lambda function, which launches the RNA-Seq analysis workflow with the following input parameters:
- Sample sheet
- GEO ID
- Other relevant metadata
- Our RNA-Seq data analysis is run with HealthOmics and the associated sequence store. We use Step Functions to launch this workflow and ingest the relevant files to the sequence store.
- Upon workflow completion, HealthOmics writes the output data (BAM files) to the output S3 bucket.
Implementation considerations
Dataset preparation
The Step Functions state machine orchestrates the ingestion of FASTQ files through the following steps:
- The state machine invokes the Map state in Step Functions that uses dynamic parallelism for increased scale, with the SRR IDs array as input. You can now launch multiple AWS Batch jobs in parallel to ingest the FASTQ files that correspond to the SRR ID input.
- The state machine checks our ingestion DynamoDB table to see if the corresponding SRR ID has already been processed and has ingested the corresponding FASTQ files. If the SRR ID ingested the files, the state machine writes the sample sheet to the FASTQ S3 bucket and terminates successfully.
- The state machine uses the NCBI-provided sra-tools Docker container and
fasterq-dumpcommand to ingest the FASTQ files. The state machine generates the set of ingestion commands and starts the AWS Batch job. The ingestion commands are a set of shell commands that interact with NCBI for downloading FASTQ files. These commands compress the files with pigz, and then uploads them to an S3 bucket. - The state machine updates the DynamoDB table with the ingestion status.
- If the ingestion is successful, then the state machine continues to step 5.
- If the ingestion isn’t successful, the state machine writes a message to Amazon Simple Notification Service (Amazon SNS) to notify scientists of the failure.
- A Lambda function generates the RNA-Seq sample sheet with the combined samples to analyze. This sample sheet is a CSV file containing:
- The paths to the ingested FASTQ files.
- The names of each corresponding SRR ID as input to the RNA-Seq workflow.
- The state machine notifies that the ingestion job is complete by publishing a message to an Amazon SNS topic before terminating itself.
Figure 2 provides a detailed overview of the state machine.

Figure 2. RNA sequencing data ingestion
Dataset analysis
A Lambda function divides the RNA-Seq sample sheet in compliance with the Step Functions service quota. This enables parallel processing using a Map state.
Our transcriptomic analysis workflow does the following:
- Checks if samples are single-end (one FASTQ file per sample) or paired-end (two sets of FASTQ files per sample).
- Ingests the appropriate set of FASTQ files into the HealthOmics sequence store.
- Monitors the status until all files are imported.
In parallel, a Lambda function initiates the HealthOmics RNA-Seq workflow.
Upon successful completion, HealthOmics stores the output data in Amazon S3. Finally, our state machine imports the output BAM files into the HealthOmics sequence store for future use.
Figure 3 provides a detailed overview of our state machine.

Figure 3. RNA sequencing workflow
Cleanup (optional)
Delete all AWS resources that you no longer want to maintain.
Conclusion
HealthOmics removes the heavy lifting associated with gaining insights from genomics, transcriptomics, and other omics data. We used RNA-Seq analysis to showcase an example scientific workflow that can benefit from HealthOmics. When using HealthOmics in combination with Step Functions, scientists can automate the entire workflow from initial dataset preparation to archival. To learn more, we encourage you to explore our HealthOmics tutorials on GitHub.
Related information
- Genomics workflows, Part 1: automated launches
- Genomics workflows, Part 2: simplify Snakemake launches
- Genomics workflows, Part 3: automated workflow manager
- Genomics workflows, Part 4: processing archival data
- Genomics workflows, Part 5: automated benchmarking
- Genomics workflows, Part 6: cost prediction
Author: Rostislav Markov